| IV. Genetics of physiological
traits and others
15.The feasibility of PCR-based allele mining for stress tolerance genes
in rice
L. RANGAN1,2, S. CONSTANTINO1,
G.S. KHUSH1, M.S. SWAMINATHAN2
and J. Bennett1 1)International Rice Research Institute, Manila, Philippines 2)M.S. Swaminathan Research Foundation, Taramani, Chennai, India The International Rice Genome Sequencing
Project (IRGSP) aims to complete the sequencing of the genome of Oryza
sativa L cv. Nipponbare within 10 years. One of the many applications of
this sequence information will be to devise rapid and inexpensive PCR strategies
to isolate useful alleles of rice genes from a wide range of rice cultivars
and related species and genera. This capability will be important for giving
rice breeders direct access to key alleles conferring (1) resistance to
biotic stresses, (2) tolerance of abiotic stresses, (3) greater nutrient
use efficiency, (4) enhanced yield, and (5) improved quality, including
human nutrition. This approach to allele mining is already available on
a limited scale because of the approximately 105 rice entries already existing
in public sequence databases but will be greatly enhanced when IRGSP provides
both the sequence and the physical map location of each Nipponbare gene.
Cultivated rice and its relatives
provide an opportunity to test the evolutionary range
over which PCR-based allele mining can be successful. Nipponbare
is a japonica cultivar and therefore belongs to isozyme group VI of 0.
sativa (Glaszmann 1987). For each gene, the Nipponbare allele will be most
closely related to the alleles of other japonica cultivars and then progressively
less closely related to alleles of (1) the isozyme groups I-V of 0. sativa,
(2) the other AA genome species, (3) the non-AA genome species of genus
Oryza, (4) related grass genera such as Porteresia, and (5) the other cereals.
Vaughan (1994) describes the genus Oryza and related grasses (subfamily
Oryzoideae), while Kellogg (1999) summarizes the evolutionary relationships
among the grasses in general, including the cereals. The germplasm used
in this study of allele mining comprised 64 accessions, including at least
two representatives of each of the six isozyme groups of 0. sativa, two
accessions of each of the seven other AA genome wild species, 1 or 2 accessions
of each of eleven non-AA genome wild species, one accession of each of
five closely related grass genera, and 1-3 accessions of each of five other
cereals. Genomic DNA was extracted from each accession for amplification
by PCR primers.
The evolutionary distance over which
PCR-based allele mining succeeds will depend strongly on the location of
the PCR primers within the gene. If the primers are located at highly conserved
sequences within the coding region of the gene, amplification will occur
over the widest evolutionary range but can hardly be described as allele
mining, because the amplicon will be merely an internal fragment of the
gene and devoid of meaningful function; such primers are also likely to
lack specificity and may amplify non-alleic loci that share the conserved
motifs. As true allele mining requires that the amplicon should include
most and preferably all of the functional segments of the gene, the primers
should for preference be located upstream of the promoter and downstream
of the terminator. However, we began our evaluation of PCR-based allele
mining with a compromise location: in the 5�- and 3�-untranslated regions
immediately outside the coding region of the gene. These 5�3� primers were
expected to amplify the entire coding region of the gene along with any
introns. For each gene, we also designed a second pair of primers located
within the coding region but as close as possible to the regions encoding
the N- and C-termini of the protein. The use of the NC primers provided
a way of distinguishing between specific and non-specific priming by the
5�3� primers: when amplification was specific, the amplicon generated by
the 5�3� primers was a predictable number of base pairs longer than that
generated by the NC primers (Table 1). The number of base pairs would depend
on exactly where the 5�3� and NC primers were located, the location being
constrained by the need to satisfy two criteria: a G+C content of 50-60%
and a length of 20-25 bases.
We evaluated our protocols for allele
mining by focusing on three genes important in abiotic stress tolerance.
These genes encoded (1) calmodulin (Calmod, Z12828), (2) a late embryogenesis
abundant protein 3 (LEA3, AF046884), and (3) SalT (Z25811). Calmodulin
is a part of the network of signal transduction pathways centered on calcium
ions as second messenger in stress tolerance (Epstein 1998). LEA3 accumulates
in cells to protect them against ionic changes accompanying various stresses
(Skriver and Mundy 1991). SalT accumulates in rice leaf sheaths and roots
in response to salt and drought but its role is not clear (Claes et al.
1990). Both LEA3 and SalT are induced by ABA treatment (Moons eta!. 1995).
A common feature of the three genes
is that they are members of multigene families. Family members with very
similar sequences may have dispersed around the genome into non-allelic
locations or may have remained as tandem repeats within a single genetic
locus, confounding the usual genetic definition of allelic and non-allelic.
Our aim was to design primers giving specific amplification of particular
alleles without unduly compromising the evolutionary range over which allele
mining can be conducted. We focussed therefore on two questions: (1) Is
amplification by the 5�3� primers specific for a single allele in each
multigene family? (2) What is the evolutionary range over which allele
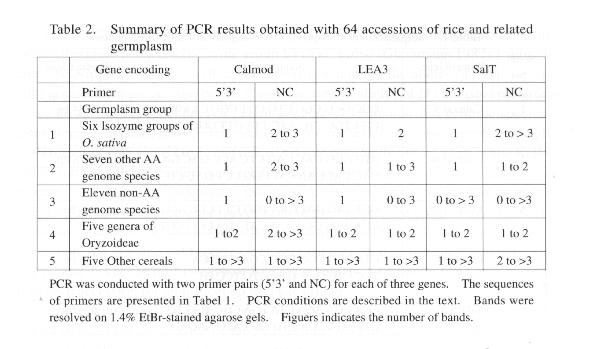
mining can be successful using 5�3� primers? Table 2 summarizes the data
obtained when genoinic DNA from the 64-germplasm accessions was amplified
using the six primer pairs listed in Table 1. The PCR components and conditions
were as follows: 50-75 ng genomic DNA, 600 nmoles of each primer, 44) cycles
of melting (94°C for 1 mm), annealing (55°C for 1 mm and 15 sec)
and elongation (72°C for 3 mm and 5 sec).
Within the six isozyme groups of
0. saliva and the seven other AA genome species, the 5�3� primers for calmodulin,
LEA3 and SalT amplified a single band, whereas the NC primers amplified
2-3 bands for calmodulin, 1-3 bands for LEA3 and 1->3 bands for SalT (Table
2). In every case, the single 5�3� band was the expected number of base
pairs larger than one of the NC bands. We conclude that, within the eight
AA genome species, allele mining using 5�3� primers is likely to be highly
effective for many genes. Primers based on 5�- and 3�-untranslated regions
are sufficiently allele-specific to avoid amplifying the additional loci
amplified by primers based on the adjacent N- and C-terminal regions and
yet are sufficiently conserved to be effective over the entire range of
germplasm for which sexual hybridization and hence the concept of allelism
are applicable in rice.
For the 11 non-AA genome wild species
examined, the applicability of allele mining proved to be variable. The
5� 3� primers for calmodulin amplified a single specific band for accessions
of 0. puncata 0. officinalis, 0. rhizoniatis, 0. eichingeri, 0. australiensis,
0. brachyantha, 0. alta and 0. grandiglumis, but not for accessions of
0. granulata and
0. meyeriana (no amplification) and 0. longiglumis (two
bands). The 5�3� primers for LEA3 amplified a single band for all 11 wild
species but in most cases this band failed to satisfy the criterion that
it should be about 100 bp larger than one of the bands amplified by the
NC primers. The 5�3� primers for SalT amplified O->3 bands and very rarely
did any of these bands satisfy the criterion that they should be about
20 bp larger than one of the bands amplified by the NC primers. We conclude
that allele mining will be successful for some non-AA genome wild species
with some genes such as calmodulin but it will not be successful for other
genes.
For the five grasses genera closely
related to the genus Oryza (Porteresia, Leersia, Hygrooryza, Luziola and
Vetiber), the 5�3� primers amplified 1-2 bands. When a single band was
amplified it appeared to be specific because it satisfied the criterion
of being 20- 100 bp larger than one of the bands amplified by the NC primers.
By contrast, for the other cereals (Lolium, Fescue, barley, wheat and maize),
multiple non-specific bands were generally obtained for both the 5�3� and
the NC primers. We conclude that PCR-based allele mining is a possibility
for some of the closely related grass genera but is unlikely to extend
to the other cereals. This is consistent with the views of Kellogg (1999)
who placed rice and the related grasses of the Oryzoideae in a subfamily
separate from the nearest subfamily, the Pooideae (to which wheat and barley
belong).
In summary, we have examined the
feasibility of allele mining of coding sequences using PCR primers based
on the 5�- and 3�-untranslated regions of genes of a reference genome such
as Nipponbare. Based on amplification of calmodulin, LEA3 and SalT genes,
our data suggests that allele mining is feasible within the eight AA genome
species of rice and for some non-AA genome species and related grass genera
of the subfamily Oryzoideae but is not feasible for other cereals, all
of which belong to other subfamilies of the grass family.
One point that needs to be checked,
however, is whether the single bands amplified by the 5�3� primers represent
unique amplicons or multiple amplicons of the same size class. There are
two occasions when multiple amplicons might be expected. The first is when
a comparatively recent gene duplication event places two slightly diverged
sequences in tandem in the genome. The second is when the genome is polyploid,
as seen with certain non-AA genome wild species. For example, 0. grandiglumis
contains the CCDD genome and yet gives a single specific band with the
5�3� primers. This band is likely to contain an amplicon from both the
C and the D genomes. We are currently exploring the most efficient means
of distinguishing between unique and multiple ainplicons of one size class.
It is interesting that 0. longiglumis (HHJJ) is the only non-AA genome
species to give two bands with the calmodulin 5�3� primers. It is possible
that the bands originate respectively from the H and J genomes. By contrast,
0. granulata (GG) and 0. meyeriana (GG) both fail to give any band with
the same primers. This result could be due to the fact that, of all the
non-AA genome species, the GG genome species are considered to be the most
highly diverged from the AA genome species (Aggarwal et al. 1997).
Within 0. sativa and even within
the AA genome species of the genus Oryza, a strict genetic meaning can
be given to allelism because cultivars are all diploid, are adequately
sexually compatible to hybridize and appear to have collinear genomes.
By contrast, the non-AA genome species are not sexually compatible, gene
transfer to 0. sativa requires embryO rescue, and introgression lines exhibit
random integration of small fragments of non-AA genomes. Although the conventional
meaning of allelism cannot be easily attached to comparisons between 0.
sativa and non-AA genome species, the value of gene isolation from non-AA
genome species is clearly established by the range of traits that have
already been transferred toO. saliva by wide hybridization and embryo rescue
(Brar and Khush 1997). In those cases where the direct PCR mining described
here cannot be used, and where screening of X and BAC libraries is too
expensive and time-consuming, other PCR techniques may be feasible, such
as PCR walking (Devic eta!. 1997). We are currently examining the feasibility
of mining complete alleles (i.e., including promoter and terminator) by
the same direct PCR approach described here. However, we suspect that the
evolutionary range over which this approach is useful for complete alleles
will be more limited than found here for complete coding sequences. It
is likely that PCR walking will be especially useful for mining of complete
alleles.
Acknowledgement This research was supported in part by a grant from the Technical
Advisory Committee of the Consultative Group on International Agricultural
Research.
Table 1. Sequences of PCR primers for amplification of three
genes
Aggarwal, R.K., D.S. Brar, N. Huang and G.S. Khush, 1997.
Two new genomes in the Oryza complex identified on the basis of molecular
divergence analysis using total genomic DNA hybridization. Mol. Gen. Genet.
254: 1-12.
Brar, D. S. and G.S. Khush, 1997. Alien introgression in
rice. Plant Mol. Biol. 35: 35-47.
Claes, B., R. Dekeyser, R. Villarvoel, M.Van den Buicke, M.V. Montagu and A. Caplan, 1990. Characterization Devic, M., S. Albert, M. Delseny andTJ. Roscoe, 1997. Efficient PCR walking on plant genomic DNA. Plant Physiol. Biochem. 35: 331-339. Epstein, E., 1998. How calcium enhances plant salt tolerance.
Science 280: 1906-1907.
Glaszmann, J.C., 1987. Isozymes and classification of Asian
rice varieties. Theor. AppI. Gent. 74: 21-30.
Kellogg, E.A., 1999. Relationships of cereal crops and other
grasses. Proc. Natl. Acad. Sci. USA 95:20052010.
Moons, A., G. Bauw, E. Prinsen, M.V. Montagu and D.V. Straeten,
1995. Molecular and physiological responses to abscisic acid and salts
in roots of salt-sensitive and salt-tolerant indica rice varieties. Plant
Physiol. 107: 177-186.
Vaughan, D., 1994. The wild relatives of rice. A genetic
resources handbook p. 3-5.
Skriver, K. and J. Mundy, 1991. Gene expression in response
to abscisic acid and osmotic stress. Plant Cell 2:
503-512.
|
")
")
")
")
")
")
")
")

")





